ニュース
高分子相溶性を予測・理解するための量子化学計算・深層学習統合解析 オープンソースプラットフォームを開発
2023年7月13日
高分子相溶性を予測・理解するための量子化学計算・深層学習統合解析
オープンソースプラットフォームを開発
三菱ケミカルグループ※1(以下「三菱ケミカル」)と統計数理研究所(以下「統数研」)との共同
研究部門「ISM-MCC フロンティア材料設計研究拠点」※2 の研究チームは、ポリマー・溶媒系の相溶
性※4 を高精度に予測する新たな手法を開発し、この研究成果をまとめた論文が米国時間 7 月 10 日に
Macromolecules 誌(アメリカ化学会)に掲載されたことをお知らせいたします(掲載論文の URL=
https://doi.org/10.1021/acs.macromol.2c02600)。
統数研と三菱ケミカルは、ポリマーと溶媒の相溶性を表す相互作用パラメータ(χ(カイ)パラメ
ータ)を高精度かつ迅速に予測する量子化学計算・機械学習統合プラットフォームを開発しまし
た。研究チームは、実験系由来の系統バイアスを持つ限られた実験データから高精度な予測モデル
を得るために、三菱ケミカルが保有するハイパフォーマンスコンピューターを用いて生成した量子
化学計算の大量のデータを利用しました。マルチタスク学習という手法を用いてこれらのタスクを
同時に学習することで、従来の機械学習のモデルに比べてより広範囲のポリマー・溶媒に適用可能
な予測モデルの構築に成功しました。さらに、開発されたモデルは、量子化学計算に基づく従来法
に比べて約 40 倍の速さで χ パラメータを計算できることが確認されました。
ポリマーの溶媒への溶解は、プラスチックのリサイクルのほか、ポリマーの合成、精製、塗装、
コーティングなど材料開発の様々な場面で欠かせないプロセスです。したがって、本研究成果は産
学の様々な課題解決に貢献できることが期待されます。また、マテリアルズ・インフォマティクス
分野におけるオープンイノベーション・オープンサイエンスの促進に貢献するため、開発したソー
スコードとデータの一部を無償で公開しました(公開 URL= https://github.com/yoshida-
lab/MTL_ChiParameter)。
※1 三菱ケミカルグループは、三菱ケミカルグループ株式会社とそのグループ会社の総称です。
※2 https://www.m-chemical.co.jp/news/2019/__icsFiles/afieldfile/2020/02/18/190610-2.pd
以上
お問合せ先
三菱ケミカルグループ株式会社
コーポレートコミュニケーション本部
メディアリレーション部
TEL:03-6748-7140
大学共同利用機関法人
情報・システム研究機構 統計数理研究所
運営企画本部企画室 URA ステーション
TEL: 050-5533-8580、E-mail: ask-ura@ism.ac.jp
研究の背景
ポリマーの溶媒への溶解は、プラスチックのリサイクル、ポリマーの合成、精製、塗装、コーテ
ィングなど、材料開発において欠かせないプロセスの一つです。例えばリサイクルでは、異種プラ
スチックが混ざったプラスチックゴミに対して、溶媒を添加することで特定の材料だけを選択的に
分離します。あるいは溶媒を相溶化剤と呼ばれる材料として用いて高機能なポリマーブレンドを作
製します。このように、ポリマーと溶媒の相溶性を予測することは学術的にも産業的にも重要な課
題です。
現在の計算化学の技術では様々なポリマー・溶媒系の相挙動を正確に予測することは難しいこと
が知られています。高分子溶液の熱力学的性質を記述するフローリー・ハギンズ理論※3 によれば、
温度・体積分率・分子鎖⾧が与えられると、高分子溶液の混合自由エネルギー※4 は、ポリマー・溶
媒間の相互作用を表す χ パラメータと呼ばれる量により決定されます。χ パラメータの予測にはポリ
マーと溶媒の溶解度パラメータの距離に基づく経験的な予測手法が最も広く用いられています。例
えば、ハンセン溶解度パラメータ(HSP)は、分子を分散項、極性項、水素結合項からなる 3 次元
ベクトルで表します。ポリマーと溶媒の相溶性は、HSP ベクトルの間の距離に基づいて推定されま
す。様々な分子の溶解度パラメータが実験的に測定されてきましたが、溶解度パラメータが未確定
の分子に対しては、原子団寄与法※5 のような経験モデルを適用して溶解度パラメータを推定しま
す。しかしながら、このような経験モデルは、特定の分子種以外の予測精度が非常に低いことが知
られています。また、量子化学計算を用いた COSMO-RS 法※6 という分子シミュレーションでも χ
パラメータを推定できますが、量子化学計算は計算時間がかかるため例えば溶媒候補分子の大規模
スクリーニング等に適用することは困難です。また、予測精度も高くはありません。さらに機械学
習による予測モデル構築のためには学習データセットが量的に不足し、実験系の性質上重大な偏り
を持つことが知られています。このような課題を解決するために、ISM-MCC フロンティア材料設計
研究拠点の研究チームは、マルチタスク学習と呼ばれる手法を用いて、量子化学計算の大量のデー
タと限られた実験データを統合的に解析することで、広範囲なポリマー・溶媒の組に適用可能な高
精度予測モデルの構築を試みました。
研究内容と成果
モデルの学習には、46 種類のポリマーと 140 種類の溶媒分子からなる 1,190 ポリマー・溶媒ペア
の χ パラメータの実験値を用いました。データセットには、温度や組成の違いに対する χ パラメー
タの測定値も含まれています。データセットのポリマー・溶媒の分子種は、化学空間全体のごく限
られた領域に分布しています(図 1 左)。またある実験系では、非相溶状態のポリマー-溶媒系の χ
パラメータを測定することが困難であるため、データの分布に大きな偏りが生じます(図 1 右)。し
たがって、このデータセットのみを用いて学習されたモデルは、一般に予測の適用範囲が狭く、非
相溶状態の χ パラメータの予測が不得意です。
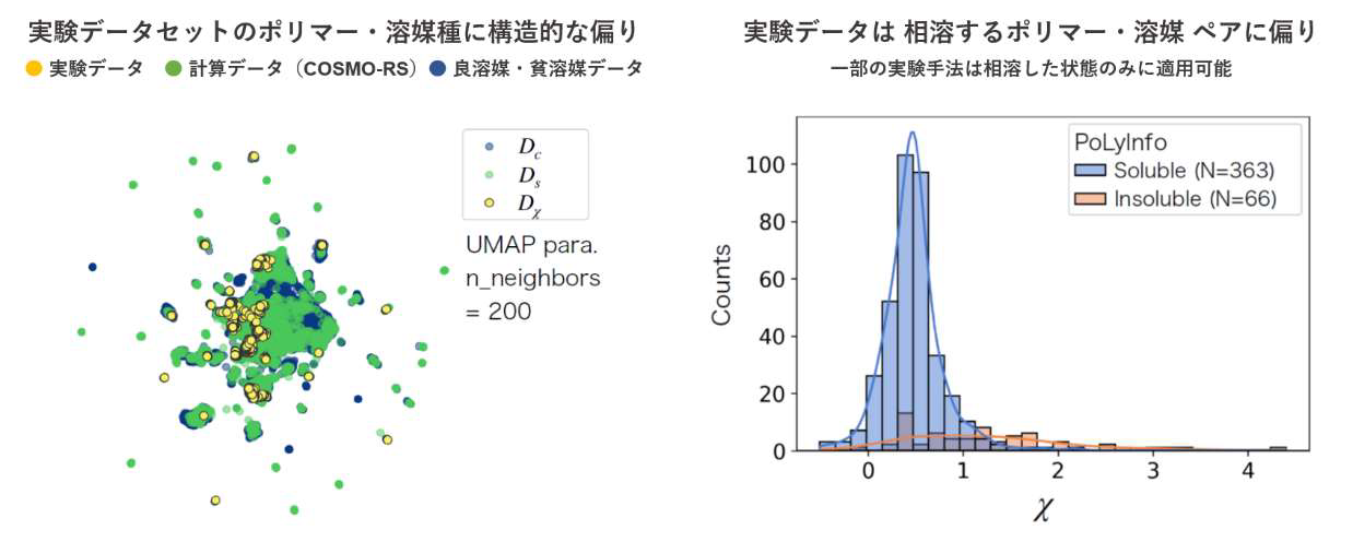
図 1. χパラメータの実験データに存在するポリマー・溶媒種には重大なバイアスが存在する。 |
この問題を解決するために、量子化学計算に基づく COSMO-RS 法を用いて、9,129 ポリマー・溶 媒ペアの χ パラメータのデータセットを生成しました。また、29,777 種類のポリマー・溶媒の組み 合わせについて溶媒が良溶媒であるか貧溶媒であるかの実験結果を表す二値ラベルが付与したデー タセットを作成しました。これら三つのデータセットを用いて、ポリマーと溶媒の化学構造から χ パラメータの実験値と COSMO-RS 法で得られた値、ポリマーと溶媒の溶解性を表す二値ラベルを 予測するディープニューラルネットワークを学習しました(図 2)。この方法はマルチタスク学習と
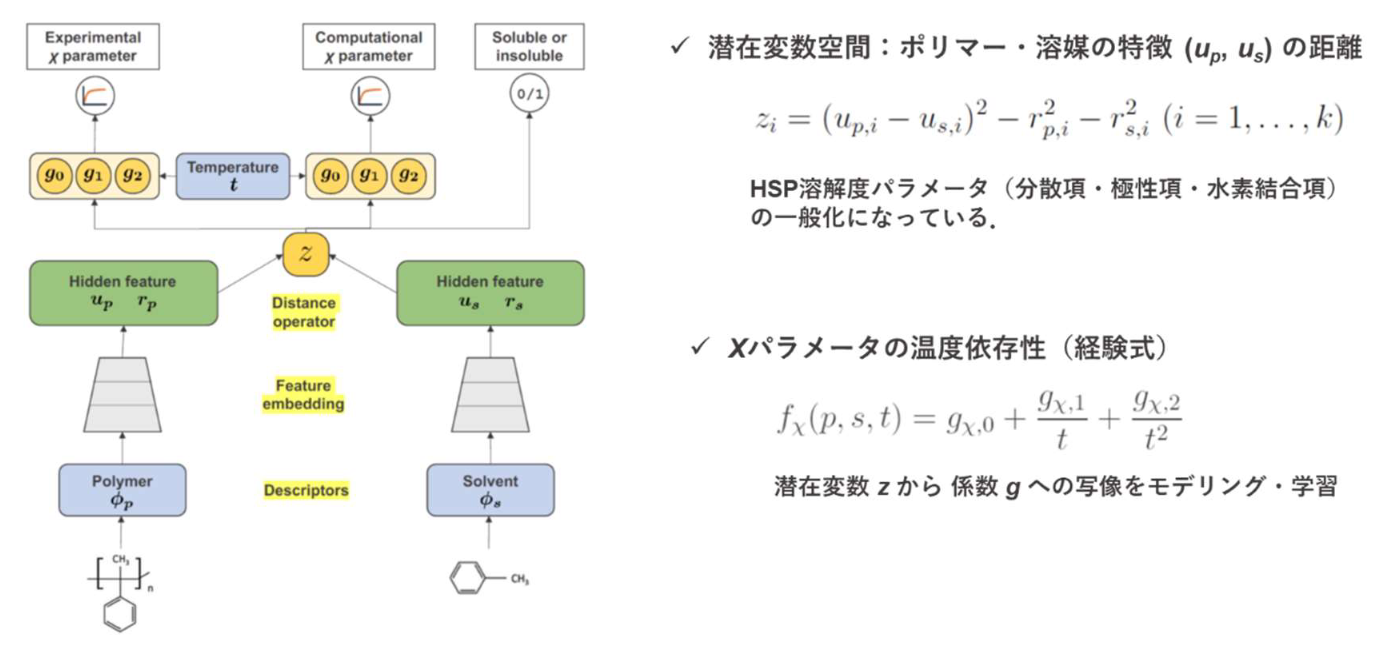
図 2. マルチタスク学習に用いたディープニューラルネットワーク |
呼ばれます。マルチタスク学習では、背後に共通のメカニズムが存在する異なるタスクを統一的な
モデルで同時に学習します。主タスクの χ パラメータの実験データは量的に限られており、実験系
由来のバイアスも含まれています。そこで、二つの補助タスクを定めて、広範囲な分子種を包含す
るデータを学習に用いることで、予測モデルの適用領域を拡大することができました。また、この
モデルは、量子化学計算に基づく従来法と比較して約 40 倍の速さで χ パラメータを計算できること
が確認されました。
このモデルは、異なる三つのタスクのいずれにおいても、非常に高い予測性能を有することが実
験的に確認されました。また、COSMO-RS 法による量子化学計算や HSP に基づく経験的手法をは
るかに凌ぐ予測力を示しました(図 3)。このモデルのアーキテクチャは、HSP のコンセプトを拡張
するように設計されています。HSP は、分子の潜在的溶解性は分散力、極性、水素結合の強さで決
まると仮定しています。一方、機械学習のアルゴリズムは、分子の溶解性には 34 種類の因子が関与
していることを示唆しています。そのうちのいくつかの因子は、HSP の 3 因子に対応していること
が分かりました。このことは、ポリマー・溶媒の相溶性決定のメカニズムには、HSP では無視され
てきた未知の因子が存在することを示唆しています。
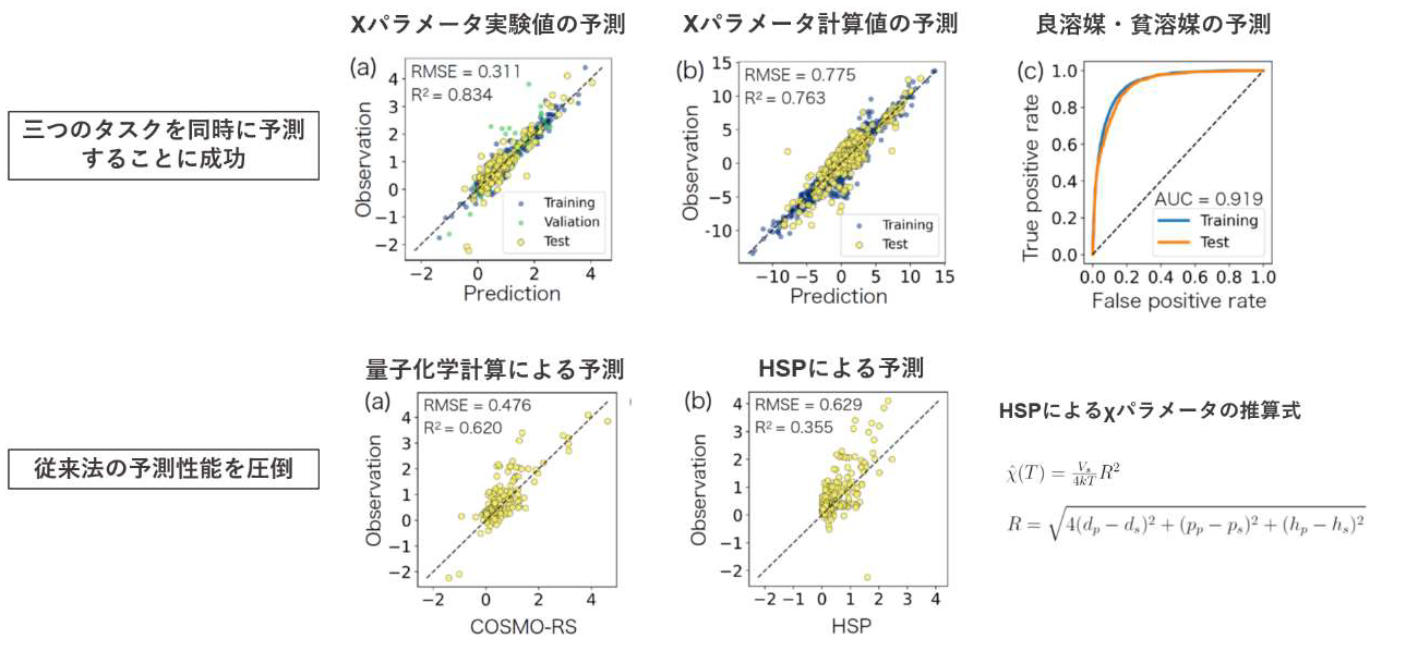
図 3. マルチタスク学習の予測モデル、量子化学計算、HSP に基づく経験モデルの予測性能の比較 |
今後の展開
今回の研究により、ポリマー・溶媒系の相溶性予測モデルを構築するための量子化学計算・深層
学習統合解析プラットフォームが完成しました。今後もデータ生産を継続しながら、モデルの予測
性能を向上させていく予定です。
ポリマーと溶媒の相溶性を予測・理解することは、今後の材料開発において益々重要になってい
きます。特に近年は脱炭素社会の実現に向けて廃プラスチック資源循環のための技術革新に対する
期待が急速に高まっています。廃プラスチックのリサイクル比率を向上させるには、様々な異種ポ
リマーに対する相溶化剤の開発が必要不可欠となります。ISM-MCC フロンティア材料設計研究拠点
は、今回のモデルを相溶化剤開発に実践展開していきます。また、機械学習技術のさらなる改善と
拡張、マテリアルズ・インフォマティクス分野におけるオープンイノベーション・オープンサイエ
ンスを促進するために、開発したソースコードとデータの一部を一般公開しました。
掲載論文
論文題目: Multitask machine learning to predict polymer-solvent miscibility using Flory-Huggins
interaction parameters
著者: Yuta Aoki, Stephen Wu, Teruki Tsurimoto, Yoshihiro Hayashi, Shunya Minami, Okubo
Tadamichi, Kazuya Shiratori, Ryo Yoshida
雑誌: Macromolecules
DOI: 10.1021/acs.macromol.2c02600
掲載日時: 2023 年 7 月 11 日 9 時(米国時間 10 日 20 時)
用語解説
※3 1942 年にフローリーとハギンズにより独立に提案された格子モデルをもとにした統計熱力学理論。現在でも高分子溶液や高分子混
合系の熱力学的性質を議論する際によく用いられる。
※4 2成分を混合することによる自由エネルギーの変化。
※5 ある分子構造を、CH3 や OH などの原子団に分けて、各原子団の寄与から、未知の構造の物性を推算する手法。
※6 量子化学計算により求まる表面電荷分布から、溶液中の分子間相互作用を評価し、活量係数・溶解度などの熱力学物性を推算する手
法。
三菱ケミカルグループ株式会社 ホームページはこちら
情報・システム研究機構 統計数理研究所 ホームページはこちら




